Genetic Testing for Beta Thalassemia
Sep 18, 2015
9891 Views
A couple planning their first baby is worried about passing on the family “lineage” of Beta thalassemia. A worried father watches his young son fumble with a cricket ball and critically wonders how much longer. An expecting mother is in a dilemma when her anaemic daughter asks her if the baby will keep her company during her next “medicine-time” at the hospital.
Beta-thalassemia (also known as Cooley’s anaemia) and sickle cell anaemia are prominent haemoglobinopathies (“abnormal haemoglobin”) in India. These are inherited disorders which pose a lifelong burden to affected individuals, especially thalassemia majors — patients with two faulty HBB genes. Individuals with one faulty gene are termed thalassemia minors and typically do not manifest symptoms (some cases are mildly affected, but do not require medical treatment).
*Thalassemia is mainly manifested as anaemia – lifelong transfusions (blood transfer through a needle) are required in severe cases.
Social factors that are barriers for reducing the burden of disease
- Kinship system, greater “value” attached to “blood marriages”— consanguinity increases the chances for offspring with inherited disorders.
- Lack of public awareness, lack of access to screening procedures.
- Absence of policies for premarital screening — currently, Indian couples do not seriously weigh the negative outcomes of carriers marrying other carriers.
Beta thalassemia – Indian scenario:
- A significant percentage (3.3%) of Indians are beta-thalassemia carriers.
- Geographical variation in India: Beta-thalassemia carriers are prevalent in Sindhis and Punjabis from Northern India, Gowdas and Lingayats from Karnataka, and sickle-cell anemia (HbS) in tribal populations of North-East India, Kerala.
Genetic testing benefits
Genetic testing must be combined with expert counseling and conventional techniques (for full confirmation of disease) to offer the following benefits:
- Screening for at-risk couples (prenatal diagnosis).
- Informed decisions about pregnancy- choice of termination in case of confirmed diagnosis (affected fetus).
- Identification of carriers in the family – untested siblings, relatives etc (“minor” cases appear normal).
Options:
- Mutation analysis for well-known variations (HBB) – these include commonly found variations in Indians.
- Complete gene analysis (HBB)
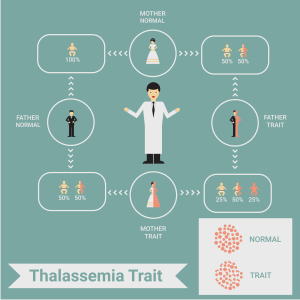
We offer the following tests:
- Beta Thalassemia Mutation Analysis: This test analyses DNA (extracted from whole blood) for the major mutations found in the HBB gene which represent the mutation spectrum found in Indian thalassemic subjects-(IVS 1-5 G>C, IVS 1-1 G>T, 619 bp deletion, Codon 15 G>A, Codon 30 G>A, FS 8/9 +G, FS 41/42 – CTTT). Molecular analysis is done via PCR (amplification of DNA) and sequencing. These polymorphisms account for >90% of the beta thalassemia cases studied in our population. Screening for these mutations plays an important role in clinical diagnosis (e.g., prenatal screening, carrier testing). Genetic testing combined with expert counseling is essential to reduce the burden for this critical disease.
- Beta Thalassemia Full Gene Sequencing (HBB): The HBB gene is analyzed by PCR and sequencing of both DNA strands of the entire coding region and the highly conserved exon–intron splice junctions.
About the Author

References:
1.Yaish HM. Thalassemia. http://www.emedicine.com/PED/topic2229.htm Accessed 6th August 2007.
2.Verma IC, Choudhry VP, Jain PK. Prevention of thalassemia: A necessity in India. Indian J Pediatr 1992; 59: 649-654.
3. Verma IC, Saxena R, Kohli S. Past, present & future scenario of thalassaemic care & control in India. The Indian Journal of Medical Research. 2011;134(4):507-521.
