Celebrating World Thalassemia Day
May 08, 2018
5897 Views
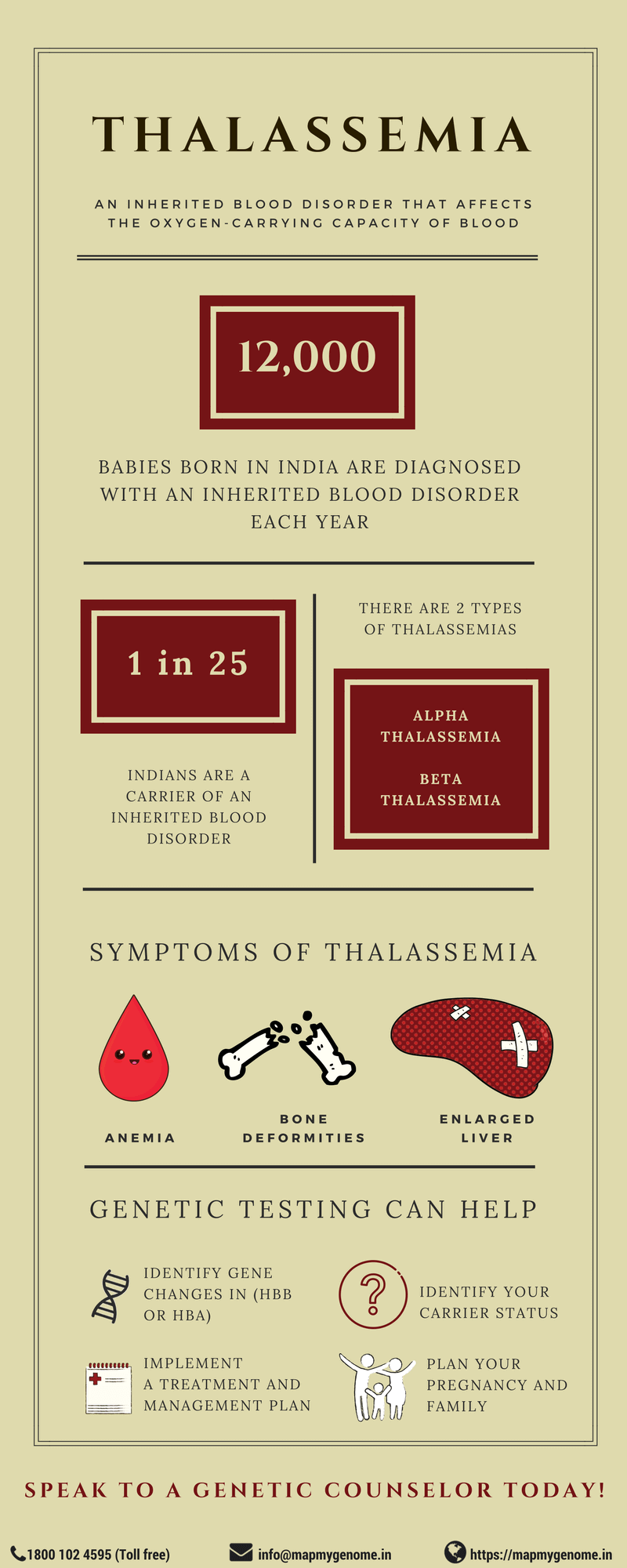
When was the last time you actively thought about the vital role blood plays in our survival? In India, approximately 12,000 babies born every year have an inherited blood disorder leading to significant health complications. Inherited blood disorders, also known as hemoglobinopathies, are a group of inherited blood disorders that affect the production of normal hemoglobin. Hemoglobin is a protein found in red blood cells (RBCs) that carries oxygen from the lungs to tissues and carbon dioxide from tissues back to the lungs.
Thalassemia, one of the hemoglobinopathies, is a genetic condition in which abnormal production of hemoglobin results in varying degree of health concerns. Thalassemia is common in many parts of the world including India, Southeast Asia, Central Asia, Mediterranean countries and Africa. In India, about 3-30% (on an average every 1 in 25) Indians are carriers of beta-thalassemia.
In order to understand the different forms of thalassemia, let us first understand the structure of hemoglobin. Adult hemoglobin is made of two alpha and two beta chains. These chains have small pockets that can contain an iron molecule that in turn binds to oxygen. Depending on which chain is damaged in the protein, Thalassemia can be of two different forms, alpha and beta thalassemia.
Alpha Thalassemia
Abnormally formed alpha chains of hemoglobin cause alpha thalassemia. It is characterized by low amounts of alpha-hemoglobin resulting in reduced levels of hemoglobin. Alpha thalassemia is clinically classified into two forms:
Hydrops fetalis syndrome/ Hb Bart Syndrome/ Alpha Thalassemia major
This is a severe form of alpha-thalassemia. When a fetus is suspected to have hydrops fetalis, ultrasound of a pregnancy 13-14 weeks presents with the following features:
- Excessive fluids in the body
- Enlarged liver and spleen
- Heart defects
- Abnormalities of the urinary system or genitals
Due to the complications in various organs, babies may be stillborn or pass away soon after birth. Pregnant mothers may also be at risk of complications in pregnancy due to hypertension, premature delivery, and abnormal bleeding.
HbH disease
It is a milder form of alpha-thalassemia. Individuals usually present with symptoms in infancy or early childhood.
- Mild to moderate anemia
- Enlargement of spleen and liver
- Yellowing of the eyes and skin
- Mild bone deformities (prominent jaw or forehead)
However, owing to varying degree of severity, some individuals may be diagnosed in adulthood or identified only during routine blood examination.
Management of alpha-thalassemia
- When hydrops fetalis is suspected and confirmed by prenatal diagnosis, early termination is considered as an option due to possible complications in the pregnancy. More studies are being conducted on therapies including intrauterine transfusions and hematopoietic stem cell transplant.
- Most individuals with HbH disease may not require any treatment. However, children and adults should be closely monitored to assess growth and normal hemoglobin levels. In extremely rare cases, individuals with severe anemia require blood transfusions.
- Iron chelation therapy may be needed for individuals with iron overload.
- Splenectomy (removal of the spleen) may be considered for individuals with enlargement in spleen due to risk for sepsis and blood clots.
- Medical attention may be required for other complications such as gallstones or leg ulcers.
Genetics of alpha- thalassemia
Alpha thalassemia is an inherited blood disorder caused due to deletion in copies of HBA1 and HBA2 genes also known as alpha-globin genes. It is inherited in an autosomal recessive pattern.
We all have two copies each, of the HBA1 and the HBA2 gene. For both these genes, we receive one copy from our mom and one copy from our dad. All four copies are involved in the production of the alpha-hemoglobin chain. Loss of some or all of these copies results in various forms of alpha-thalassemia:
- Loss of all four copies of alpha- globin genes causes Hydrops fetalis or Hb Bart syndrome
- Loss of three out of four copies of the alpha-globin gene results in HbH disease
- Loss of two out of four copies of alpha-globin genes results in alpha thalassemia trait. Most people have no or very mild symptoms as they still produce some amount of normal hemoglobin
- Individuals with loss of one of four copies of alpha-globin chain are referred to as thalassemia silent carriers. They typically have no symptoms of the disease
Inheritance of alpha-thalassemia
- When each parent is a silent carrier, meaning has three of four copies of alpha-globin gene. Their children could either have all four copies or be at risk of being a silent carrier or a thalassemia trait.
- When one parent is a silent carrier and the other parent is missing two or more copies, their child may be at risk of having hydrops fetalis, HbH disease, or having alpha thalassemia trait.
- When both parents have at least two missing copies, their children are at risk of hydrops fetalis or HbH disease.
The exact risk of the disease depends on the number of copies missing and combination of genes with the disease-causing change.
Beta Thalassemia
Beta thalassemia is characterized by low amounts of beta-hemoglobin chains that causes hypochromic anemia and reduced amounts of hemoglobin. The severity of the disease depends on the extent of imbalance between the alpha-globin chain and non-alpha globin chain. Beta thalassemia can be further classified into two types based on the severity of symptoms:
Thalassemia major or Cooley’s anemia
The clinical symptoms of thalassemia major appear between 6 and 24 months. Common symptoms include:
- Infants fail to thrive
- Grow progressively paler due to severe anemia
- Feeding problems, diarrhea, irritability, recurrent infections
- Enlarged spleen
- Bone deformities
Thalassemia intermedia
Most individuals present with symptoms in their early or late childhood. Common symptoms include:
- Pale skin and jaundice
- Liver and spleen enlargement
- Moderate to severe bone deformities
Management of beta-thalassemia
- Children diagnosed with beta-thalassemia major will require regular transfusions every 2-3 weeks to maintain minimum hemoglobin concentration for supporting normal growth and development.
- Treatment of ensuing iron overload that occurs due to its increased gastrointestinal absorption
- Bone marrow transplantation from an HLA-identical (genetically matched) sibling may eliminate the need for iron chelation therapy since iron accumulation can be managed by repeated phlebotomy (blood donation). The outcome of bone marrow transplant is based on prior risk factors such as iron load, liver fibrosis, and hepatomegaly. Children without such risk factors have higher than 90% disease-free survival rate. However, there is a small risk of rejection of transplantation.
- A possible cure can be cord blood transplantation from a related donor. It is also associated with lower risk of transplantation rejection. Couples who had a child with thalassemia can choose to have prenatal identification of HLA compatibility between the child and unaffected pregnancy. If compatible, parents may opt for the collection of placental blood at delivery for cord blood transplantation.
Complications in beta-thalassemia
Around age 10-11 years, children with thalassemia major are at risk of developing severe complications due to iron overload in the absence of iron chelation therapy. Iron overload can be monitored by regular serum ferritin concentration and in some cases, a liver biopsy. Untreated iron overload can lead to:
- Growth restriction and failure of sexual maturation in children
- Damage to heart, liver and endocrine glands due to excessive iron content
- Adults with thalassemia are at risk of osteopenia and osteoporosis
Genetics of beta-thalassemia
Beta-thalassemia is an inherited condition caused due to disease-causing changes in the HBB gene that makes the beta-globin chain. Beta-thalassemia is inherited in an autosomal-recessive manner. We all have two copies of the HBB gene. We inherit one from our dad and one from our mom. When a parent has a disease-causing change in one copy of the HBB gene, they are referred to as carriers or as having thalassemia minor. Individuals with thalassemia minor usually have no symptoms.
When both parents are carriers, they have an increased risk of having a child with beta-thalassemia. Couples who are carriers have:
- 25% (1 in 4) chance of having a child with thalassemia
- 50% (1 in2) chance of having a child who is a carrier
- 25% (1 in 4) chance of having a child that is neither a carrier nor affected
Diagnosis of alpha or beta-thalassemia
- Hematological studies: Blood smears that can identify changes in structure and amount of hemoglobin
- Hemoglobin analysis: Hemoglobin protein is made of different types of chains through different stages of life. Changes in hemoglobin chain pattern can indicate a certain type of thalassemia
- Genetic testing: Genetic testing can identify disease-causing changes or deletions in HBB, HBA1 or HBA2 gene which can confirm the diagnosis of the disease
Genetic counseling for thalassemia
Genetic testing and genetic counseling for an individual or a child suspected to have thalassemia could be beneficial for the following reasons:
- Accurate diagnosis of the disease by identifying the underlying genetic causes
- Accurate management of the disease and long-term surveillance
- Predicting risk for at-risk family members
- Couples can opt for prenatal genetic testing or preimplantation genetic diagnosis to make an informed decision about the pregnancy
- Receive psychological support to cope with the implications of the disease
Carrier screening for thalassemia
Carrier screening is a genetic test that allows the couple to learn if they are carriers for alpha-thalassemia (silent carriers or alpha-thalassemia trait) or beta-thalassemia (beta-thalassemia minor). When both parents are carriers, their children are at risk of having complications due to thalassemia.
Carrier screening is recommended for:
- Any couple with Indian ancestry planning for a pregnancy due to the high prevalence of the disease in the Indian subcontinent
- Individuals with family history of thalassemia
- Individuals opting for donor sperm or egg
Despite the high prevalence of disease in India, being diagnosed with thalassemia may be isolating for many due to lack of accessible resources. In addition, financial costs associated with treatments and need for life-long surveillance can be challenging for many families. Speaking with a genetic counselor can help you to better understand possible underlying genetic causes and discuss emotional and medical implications for yourself and your family members. A genetic counselor is a specialized health care professional who has expertise in medical genetics and psychological counseling. If you would like to set up a genetic counseling appointment CALL US on 1800 102 4595 (toll-free) or 040-66986700or WRITE TO US at info@mapmygenome.in
Resources for families with thalassemia
Support groups can be a valuable resource for individuals and families who are interested in connecting with people with similar experiences. Following are some resources if you or someone you know has been diagnosed with thalassemia:
http://www.thalassemiaindia.org
http://www.thalassemicsindia.org/index.php
Reference
Mohanty, D., Colah, R.B., Gorakshakar, A.C. et al. J Community Genet (2013) 4: 33. https://doi.org/10.1007/s12687-012-0114-0
https://www.nhp.gov.in/thalassemia_pg

