HB2
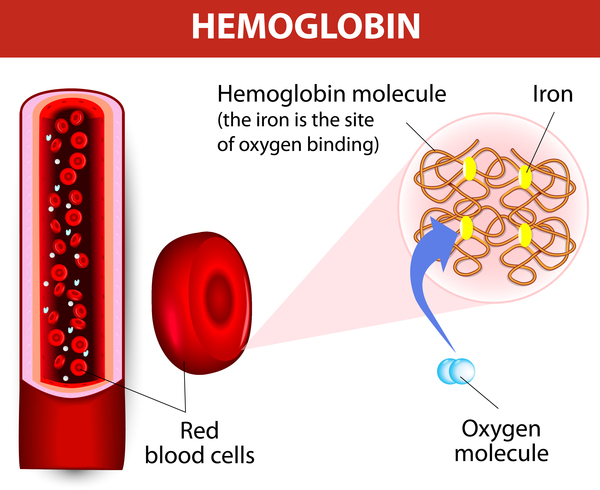
Alpha thalassemia is a blood disorder that reduces the production of hemoglobin. Hemoglobin is the protein in red blood cells that carries oxygen to cells throughout the body.
In people with the characteristic features of alpha thalassemia, a reduction in the amount of hemoglobin prevents enough oxygen from reaching the body‘s tissues. Affected individuals also have a shortage of red blood cells (anemia), which can cause pale skin, weakness, fatigue, and more serious complications.
Alpha thalassemia typically results from deletions involving the HBA1 and HBA2 genes. Less commonly, changes to the DNA sequence in or near these genes cause alpha thalassemia. Such changes are often referred to as nondeletion variants. Both the HBA1 and HBA2 genes provide instructions for making a protein called alpha-globin, which is a component (subunit) of hemoglobin.
People have two copies of the HBA1 gene and two copies of the HBA2 gene in each cell. Each copy is called an allele. For each gene, one allele is inherited from a person‘s father, and the other is inherited from a person‘s mother. As a result, there are four alleles that produce alpha-globin. The different types of alpha thalassemia result from the loss or alteration of some or all of these alleles.
Deletions and nondeletion variants in one or more alleles reduce the amount of alpha-globin cells produce. Nondeletion variants tend to reduce alpha-globin more than deletions.

{kind=link}
{kind=link}